|
|
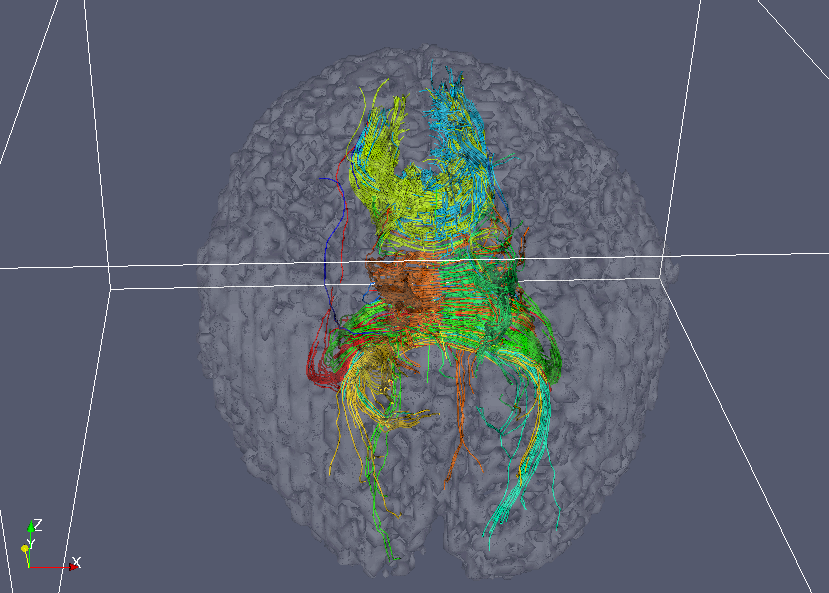
ConnectSegmentTutorialTutorial: Connectivity-based segmentation of the corpus callosumThis tutorial uses Camino to perform a connectivity-based segmentation of the corpus callosum. The methods are explained in more detail in [1]. We use a single brain from the data set of [1], which is also used in the tractography tutorial. Unlike [1], we fit a single diffusion tensor to each voxel in the brain using the standard linear reconstruction. In addition to doing the segmentation, the tutorial shows some other options to process tractography data. Reconstructing the dataWe begin with the data in voxel order in the file Step 1 is to get the tensors: The grey matter atlasThe atlas is defined in [3] and contains labels for 52 regions of the brain. The labels are defined in a T1-weighted template image from the Brainweb project. The labels were warped into the space of the subject's diffusion image by calculating a warp from the template to the subject T1, and then from the subject T1 to the diffusion image. We define a mask of the atlas, which we will use to restrict the analysis to pathways that enter the labelled regions. This is easily done in Matlab: A header is required for the mask image. Tracking from the corpus callosumBefore running a lengthy PICo process, we'll track a few streamlines to make sure that everything is set up correctly. The ROI in the corpus callosum is defined by hand in SNAP.
The call to does a couple of things. First, we use the mask as a "waypoint" image. That is, any streamlines that do not enter the mask are discarded. Secondly, we use the mask as an "exclusion" image. By default, streamlines that enter an exclusion image are discarded, but the The command converts the streamlines into VTK polylines, which we can view in Paraview. The target file is passed to We can now do the PICo tracking for real, with 1000 iterations per voxel. We'll save the results to disk, so we can do different analyses without re-running the tractography. In order to save disk space, we save each streamline as a list of voxels in the diffusion image space, and use the gzip algorithm to compress the output.
Connection probabilities to the grey matter targetsCamino offers a number of options for dealing with probabilistic streamlines. The simplest thing is to make an image of the PICo from each seed point. This is an Analyze image where the intensity in each voxel is the fraction of streamlines from a particular seed point that intersect that voxel. To do this, we run
The output is one Analyze image per seed point. The output files are named Next, we get the fraction of probabilistic streamlines that connect to each target region by specifying the atlas as a target file. The output is an Analyze image for each seed, as before, except this time the intensity of each voxel in the target file is the PICo to that target, and the intensity of all other voxels is zero. This time, we also use the mask as a waypoint file, so streamlines that do not connect to any region of interest are discarded.
Any connection probability image can be passed to to get the maximum, minimum, mean, or median value over all seed points in an ROI. We'll get the mean target connection probability over all seed points:
The result is shown below.  The command converts the target connection probabilities to a text file. The command in this example is
For each seed point, the output is Connectivity segmentation of the corpus callosumWe perform the connectivity segmentation with the command
There are two differences to the earlier commands: we specify The numbering scheme for the output is as follows: In the "labels" image, each seed point is assigned the value of the target to which it has the highest connectivity (see image below). The "labelcp" image contains the PICo for the target with the highest connectivity: the value in each seed point is the fraction of the total streamlines (1000 in this case) that connect to the target region specified in the "labels" image. The "labelcp" image gives an indication of the confidence in the target labeling (higher is better). A more detailed analysis of the labeling is possible using the output from 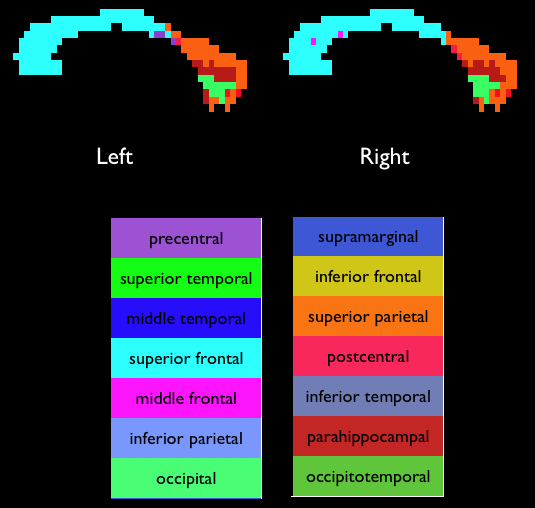 References[1] Cook P A, Zhang H, Avants B, Yushkevich P, Alexander D C, Gee JC, Ciccarelli O, Thompson A J, An Automated Approach to Connectivity-based Partitioning of Brain Structures, LNCS 3749, p. 164-171, 2005 [2] Parker G J M, Haroon H A and Wheeler-Kingshott C A M, A Framework for a Streamline-Based Probabilistic Index of Connectivity (PICo) using a Structural Interpretation of MRI Diffusion Measurements, Journal of Magnetic Resonance Imaging 18:242-254, 2003 [3] Yushkevich P, Dubb A, Xie Z, Gur R, Rur R, Gee J, Regional Structural Characterization of the Brain of Schizophrenia Patients, Academic Radiology 12:1250-1261, 2005 |